
What’s new in biology: June 2026
The most effective weight-loss drug so far, cancer breakthroughs, gene editing for cholesterol, ancestral CRISPR systems, and more.


Niko McCarty and Saloni Dattani review important things happening in the world of biotechnology and medicine.
We’ve been writing regular round ups for a little while now, but so much has happened recently that this month’s post feels like it contains a year’s worth of breakthroughs. So pour yourself something cool, get cosy, and enjoy!
First, everything new in cancer. In our round up last month, we shared the news of daraxonrasib, the new breakthrough drug that roughly doubles survival in late-stage pancreatic cancer, which has long been considered untreatable.
This past week, however, there were results from many more major cancer trials presented at the American Society of Clinical Oncology conference (ASCO 2026), and we wanted to share some highlights.1

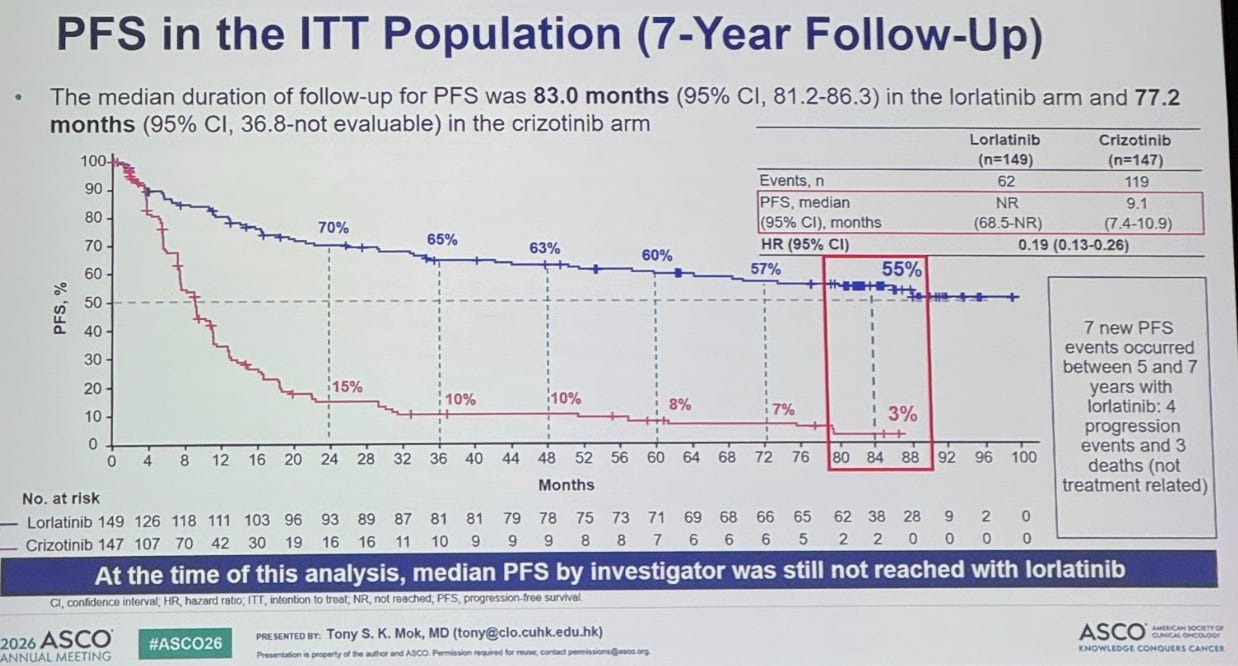
One presentation showed 7 year results for lorlatinib, a precision drug for metastatic lung cancers: specifically, a type (ALK-positive) that’s common in non-smokers. You rarely see a survival curve like it. 55 percent of patients were still progression-free after 7 years, versus just 3 percent on the older drug crizotinib.
A new prostate cancer drug, talazoparib, halved the risk of progression when added to hormone therapy, compared to hormone therapy alone, for prostate cancers carrying specific DNA repair gene mutations. Talazoparib belongs to a class of drugs (PARP inhibitors) that have already advanced treatment of breast and ovarian cancers with BRCA mutations. This trial suggests the same strategy also works for prostate cancer.

There was also long-term data on a recent endometrial cancer drug, dostarlimab, an antibody that was approved in 2023. The headline result after 4 years on the RUBY trial was that 58 percent of patients whose tumours carried ‘mismatch repair deficient’ mutations still hadn’t progressed when the drug was added to chemotherapy, versus just 16 percent on chemotherapy alone. That’s a huge difference! The drug is a ‘checkpoint inhibitor’: T cells have built-in ‘checkpoints’ that hold them back from attacking cells unnecessarily, but tumours exploit these to avoid being attacked, so blocking the checkpoint frees the T cells to attack the tumour.
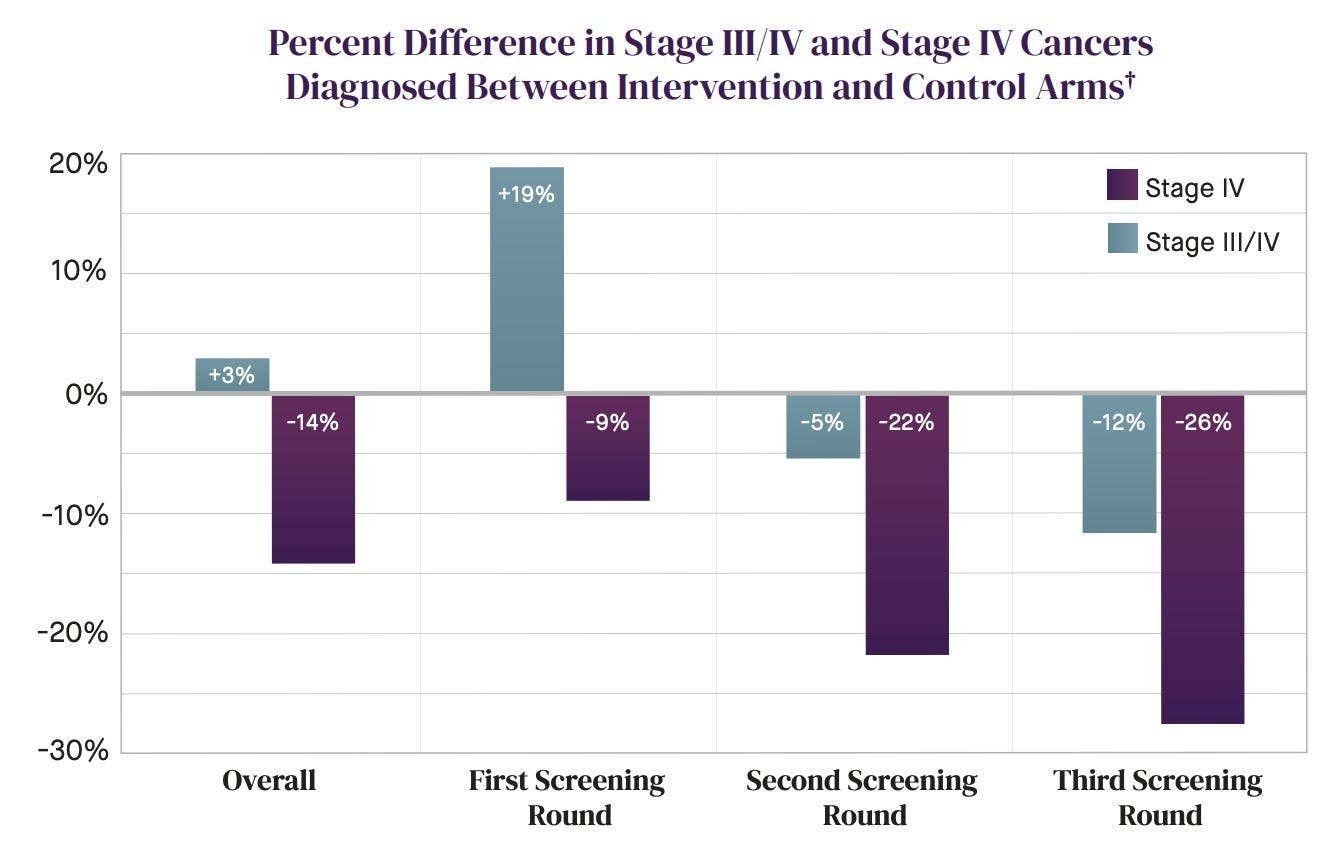
An early cancer detection blood test, the NHS Galleri blood test, was tested in a large RCT in England with about 142,000 people. The results are a little confusing, so let’s start by clarifying what a ‘good result’ here would look like: detecting non-benign cancers that would’ve missed otherwise, or catching them earlier so treatment could start sooner.
On the first count, it was successful: roughly quadrupling the overall detection rate and cutting cancers caught only at emergency presentation by around a quarter. On the second, it detected some cancers earlier: with a 16 percent rise at stage I–II, and 14 percent reduction at stage IV. But the trial missed its primary goal: there was no significant drop in combined stage III and IV cancers (because stage III diagnoses rose more than expected and roughly cancelled out the stage IV reduction). One way to interpret this – since the largest jump in detection was at stage III and the combined late-stage count barely changed – is that the ‘early’ detection test was mostly catching cancers at around stage III, rather than much earlier. That may still be useful, of course.
And finally, an mRNA cancer vaccine that might actually work. In people with high-risk melanoma, who’ve already undergone surgery, it halved the risk of cancer recurrence or death, compared to Keytruda (the blockbuster cancer immunotherapy drug) alone, in a phase 2b trial over 5 years.
Many previous mRNA cancer vaccines have been doomed by how they pick their targets. They sequence a tumour’s mutations and design the vaccine against the resulting ‘neoantigens’ (newly formed mutant proteins in a cancer cell), but most of those neoantigens are never actually displayed on the cell surface so the immune system can’t recognize them anyway. Melanoma is different: its neoantigens are readily displayed and recognize, which is why drugs like Keytruda already work well against it and why a vaccine has a much better chance of working as well.
Now, let’s move over to non-ASCO2026 news.
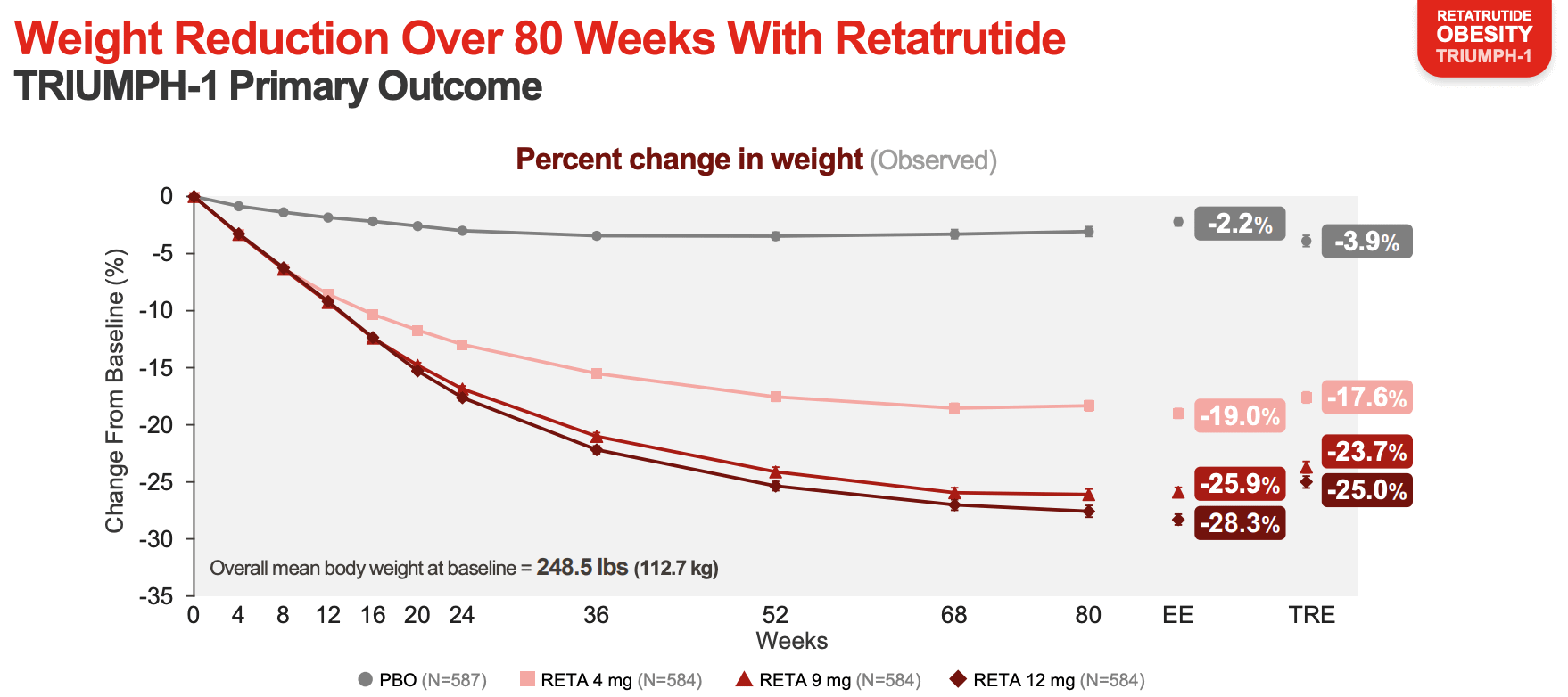
The most effective weight loss drug so far. Retatrutide, a new weight-loss drug, led to the largest reductions seen so far: roughly 28 percent loss in body weight at the highest dose in a phase 3 trial. The effect roughly matches the effects of bariatric surgery, where surgeons reduce the size of the stomach and reroute the small intestine so patients feel full earlier.
Retatrutide is an engineered peptide that acts on three receptors at once: the GLP-1 receptor, where it mimics a gut hormone that curbs appetite and slows gastric emptying; GIP, which also dampens appetite and improves how the body handles fats and sugars; and glucagon, which increases energy expenditure. It can do this because the three hormones are evolutionarily related, with similarly shaped receptors, and scientists engineered it as a single peptide that fits into all three of them.
Over the past few years, weight loss drugs have broadened in their effects: from one receptor (semaglutide) to two (tirzepatide) to three (retatrutide). More targets probably means higher efficacy in general, but it’s probably not linearly additive, with each added receptor contributing somewhat less than the last.
The first in vivo gene editing therapy. A single injection has permanently edited away a rare disease: hereditary angioedema, a rare condition where a genetic mutation causes sudden, potentially fatal swelling attacks. In a phase 3 trial, a single dose of gene editing therapy reduced attack rates by 87 percent relative to placebo, and 60 percent of patients remained attack-free entirely over 6 months.
It’s notable because it’s the first time gene editing has worked inside a living human body (in vivo) in a late-stage trial. Previous gene editing medicines like Casgevy, the first approved CRISPR drug, for sickle cell disease, worked outside the body, ex vivo, meaning they involved removing cells from the patient, editing them in the lab, and replacing them. But this new gene editing therapy packages CRISPR machinery inside a lipid nanoparticle, a tiny fat bubble, that travels through the bloodstream to the liver and edits DNA there directly.
Editing within the body will make the therapy far simpler in the real world than editing outside the body. It’s likely easier to scale up, and opens the door to treating diseases where you can’t easily remove and return cells. For now, the lipid nanoparticle delivery system mostly targets diseases of the liver, but researchers are working to extend it to other tissues like the lungs and eyes.
Permanent gene-editing to reduce cholesterol. Speaking of gene editing: a gene therapy has now been used to permanently edit a gene in liver cells to lower LDL cholesterol. These results come from a phase I trial, in which 35 adults with an inherited disease causing very high blood cholesterol (hypercholesterolemia) or premature coronary artery disease got the therapy.
The therapy, called VERVE-102, is carried by a GalNAc lipid nanoparticle, which delivers it to the liver, where cholesterol is produced. There, the therapy edits the PCSK9 gene. PCSK9 regulates cholesterol levels in the body, typically reducing how much cholesterol is removed from the blood and taken up by liver cells. The way it does this is by binding to the LDL receptor, which grabs cholesterol, and tagging it for destruction, meaning less cholesterol is absorbed from the blood. So, by editing the PCSK9 gene, LDL receptors aren’t broken down as much, more cholesterol is taken up by liver cells from the bloodstream, and blood cholesterol levels are reduced.
At the highest dose, the average reductions were huge: PCSK9 levels fell by 88 percent and LDL cholesterol fell by 62 percent. These changes lasted more than a year, with no observed toxicity at the higher doses. VERVE-102 is possibly a one-off treatment, which is great because around 40 percent of people taking cholesterol-lowering medicines stop taking them regularly.
The target of the gene edit, PCSK9, has actually become one of the most heavily pursued ones in cholesterol medicine. Several recent drugs act on it, including injectable antibodies like evolocumab, which mop up PCSK9 directly, and the oral pill enlicitide. There’s also inclisiran, an siRNA drug that reduces the liver’s production of the protein, which is off-the-shelf and needs an injection just once every six months. The patients who’d benefit most from a one-time gene edit instead of those alternatives are likely not the wider population, but rather people with inherited conditions causing high blood cholesterol, who have a strong lifelong need to reduce their cholesterol and would most value a permanent fix.
An incredible feat of pharmaceutical synthesis. One of the recent drugs that helps patients lower their cholesterol level by blocking PCSK9 is called enlicitide. In a recent phase 3 trial, it lowered LDL cholesterol by an additional 50–60 percent for people already on statins.
Enlicitide is chemically fascinating for two reasons. One is that it’s formulated as a daily pill: all the other PCSK9 drugs on the market, including antibodies like evolocumab, the siRNA inclisiran, and the gene editor VERVE-102, have to be injected. The second is that enlicitide is a synthetic peptide. That’s surprising, because peptides are normally broken down in the gut before they can work. So how can a peptide be taken orally?
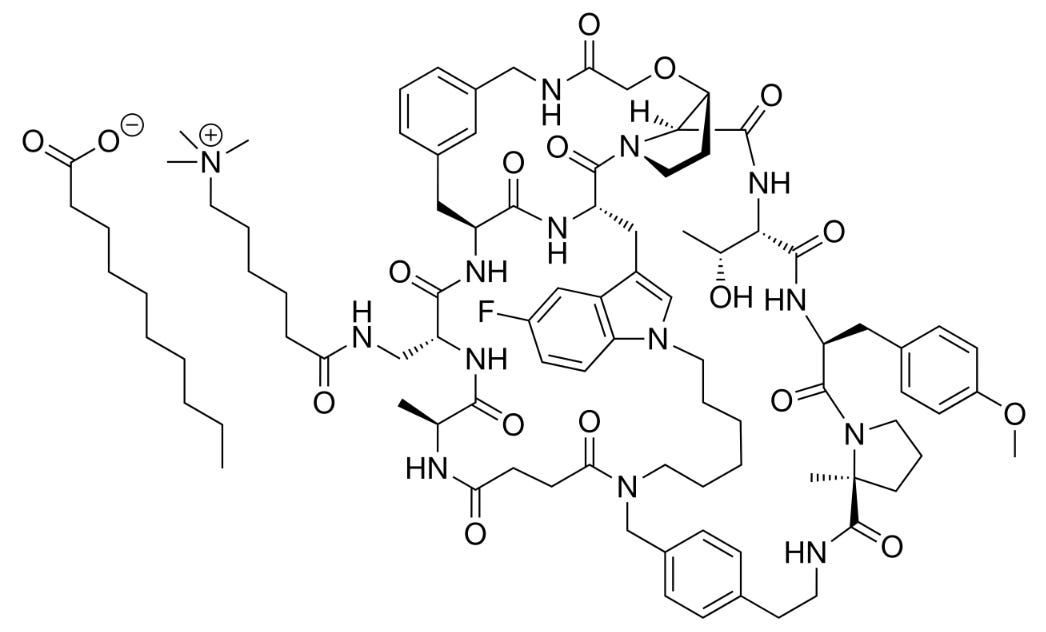
Well, enlicitide has an unusual structure. It’s a synthetic peptide made from eight amino acids that fold into a ring. This ‘closed’ shape helps the molecule survive the stomach and get absorbed in the small intestine. But the ring also makes enlicitide hard to manufacture. Some of its amino acids are ‘unnatural’, meaning they don’t appear in nature, so the peptide can’t easily be made by engineered cells, unlike other peptide drugs like semaglutide. And it’s hard to make chemically, too: the original synthesis took 63 reactions, and resulted in very low yields. The main bottleneck is the step where the chain closes into a ring, called macrocyclization.
Now, in a new study, Merck scientists improved the yield massively: with a 14-fold increase (to 70 g/L, at over 99 percent purity) by merging traditional chemistry with enzymes. They split enlicitide into three segments, called Northern, Eastern, and Western. The Eastern and Western parts were easy to make with conventional chemistry, but the Northern bit, which contains the unnatural amino acids, was instead built using enzymes. The team used directed evolution – repeatedly mutating an enzyme and keeping the best-performing ones over successive rounds – to engineer seven enzymes which, together with three natural ones, assemble the fragments and close the final ring. It’s one of the most ambitious feats of pharmaceutical chemistry ever published. And it shows that, even the most complex peptide drugs, made today at low yields, could in principle be manufactured at industrial scale through clever enzyme engineering.
A cure for some patients with hepatitis B. Hepatitis B is a virus that causes liver damage and liver cancer in middle- and old-age. It’s preventable with birth-dose hepatitis B vaccines, but there are still around 250–300 million people globally who have chronic infections, mostly being infected before the vaccines were introduced.
Hepatitis B infections are particularly difficult to ‘cure’ because the virus has a little mini-chromosome that goes inside the nuclei of liver cells. So even if you destroy all the RNA transcripts, the mini-chromosome lives on and the infection persists.
But new drugs could destroy them. In two phase 3 trials, about 1,800 adults with chronic hepatitis B infections were treated weekly for 24 weeks with antisense oligonucleotides (short strings of nucleotides, called ASOs) or placebo. These ASOs bind to the virus’ RNA transcripts and mark them for destruction by enzymes. Then, the ASOs are recycled by the body.
Interestingly, even though the ASOs target RNA, they also reduce viral DNA, because hepatitis B can only make new DNA by reverse transcription (turning its RNA back into DNA), so cutting off the RNA cuts off the supply of new DNA. It also stops the virus churning out viral proteins like HBsAg, a surface antigen produced in enormous quantities to misdirect our immune cells.
About 20 percent of the patients who received the drug, called bepirovirsen, had a ‘functional cure,’ versus none in the placebo group. Here, a functional cure meant no detectable hepatitis B viral DNA and a loss of HBsAg sustained for at least 24 weeks post-treatment.
Human cells can ‘swap’ DNA with each other. It’s long been assumed that each human cell’s genome is sealed up in the nucleus and evolves independently of its neighbours. A new study finds that isn’t always true. The researchers found that large, chromosome-sized pieces of DNA can move directly from one human cell into another, lodge in the next cell’s genome, and remain functional, getting transcribed into RNA and translated into protein.
How on earth are they doing it? The authors observed DNA passing through ‘nanotubes’, which are thin bridges that briefly form when cells touch. Scientists have known about these for a few decades, and that cells were shuttling things like mitochondria through them, but it wasn’t confirmed that genomic DNA could be transferred through them until now.
To measure it, the researchers grew two batches of human cells together, and tagged DNA packaging proteins in each batch with a different fluorescent colour, one green and one red. They then stressed the cells to break their chromosomes or scatter them during cell division (using mitosis-blocking drugs, radiation, and CRISPR) and filmed what happened. On the time-lapses, they watched DNA of one colour travel down a nanotube into a cell of the other colour.
It’s not yet clear how much this matters in the body. But the authors note it could let genomically unstable cancer cells pass on their mutations to neighbouring cells. If that’s the case, the nanotubes themselves could be a target for cancer therapies.
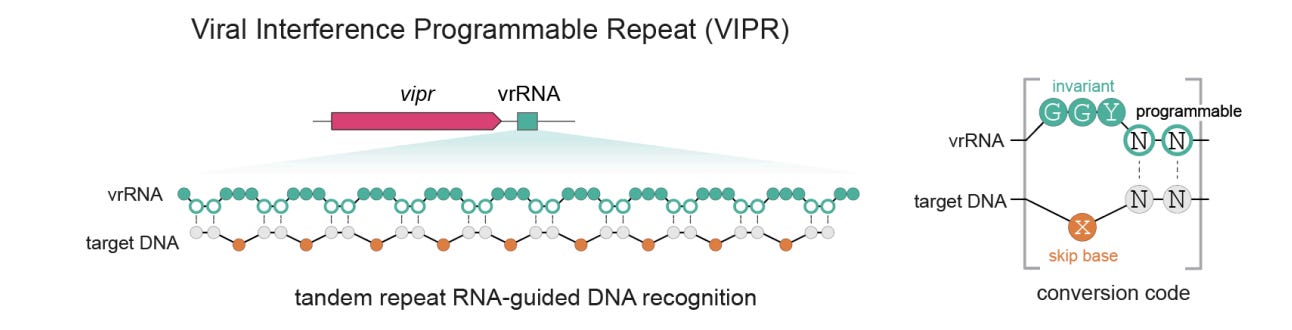
CRISPR’s ancestors, hiding in bacteriophages. Researchers in Jennifer Doudna’s lab at UC Berkeley, which helped pioneer CRISPR as a gene-editing tool, have found a system that predates CRISPR and described it in two new preprints. CRISPR is a defense system bacteria use to recognize and cut up invading DNA, including from viruses. A subset of its proteins are built around a core structure called a RAMP, which has stayed remarkably constant over many millions of years, even as the genes encoding it have drifted apart. So the team used AlphaFold, the protein structure prediction tool, to hunt for proteins that look like RAMPs but are encoded by unrelated sequences. They found some hidden in bacteriophages, probably as a weapon that they use against rival phages.
They named the system VIPR, and it appears to be older than CRISPR itself. Like CRISPR, it’s guided by an RNA, but it differs in a few notable ways. It doesn’t cut DNA at all; instead it clamps onto a target sequence and silences the gene by physically blocking the enzymes that read it, leaving the DNA unaltered. It also finds that target with an unusual ‘noncontiguous’ code: the guide RNA reads the DNA two bases at a time and skips every third one, wrapping around the intact double helix rather than unzipping it. And it’s tiny: Vipr proteins are only about 180 amino acids, roughly an eighth the size of Cas9, paired with an RNA under 100 nucleotides. That means the whole thing can fit very easily into an AAV vector (often used as a delivery vehicle for gene therapy), and in principle you could pack many of them into a single therapy to fix several genetic errors at once.
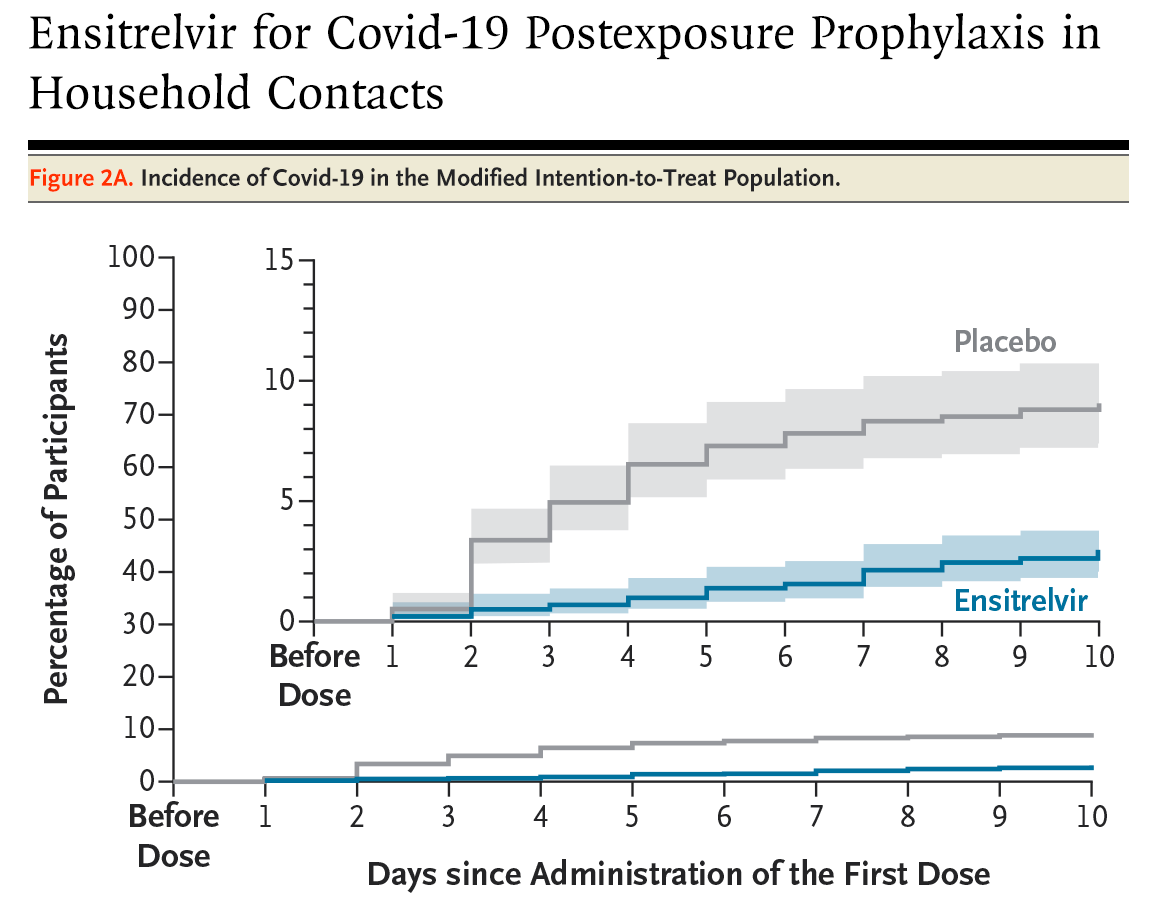
A preventive antiviral for Covid-19, finally. The first oral drug to prevent COVID after exposure succeeded in phase 3 trials. It’s called ensitrelvir and is taken as a 5-day course of pills. In a phase 3 trial, people who took it after a household member caught COVID were 67 percent less likely to develop symptoms themselves.
It’s taken a long time to get here. A preventive antiviral pill is scientifically more challenging to develop than an mRNA vaccine, which mostly needs the specific genetic sequence (of the virus’s spike protein) to be identified and swapped in. In contrast, an antiviral like this is a custom-designed molecule that physically blocks a viral protein. That’s exactly why the vaccines were so important: they could be developed quickly, at a time when they were needed most.
Unlike the vaccines, ensitrelvir works by blocking the virus’s protease, a cutting enzyme it uses to mature its proteins. Because the protease mutates far more slowly than the spike protein the vaccines target, the drug should stay effective across naturally circulating variants. It could still face drug resistance over time, but the escape mutations seen so far seem to come at a cost to the virus’s own fitness. It’s worth noting that it’s approved in the US for post-exposure prevention specifically, not as a general pre-exposure prophylactic, and not for treating active infection.
The first approved PROTAC drug. The FDA approved the first-ever ‘PROTAC’ drug, vepdegestrant, to treat a common form of advanced breast cancer. While most drugs work by blocking a disease-causing protein by binding to a pocket it uses to attach to its natural target, a PROTAC instead just destroys the protein. It’s a two-sided molecule that uses one end to grab the target protein and the other end to grab an enzyme (an E3 ubiquitin ligase) that tags the protein for recycling. This physically brings the two together, so the target protein gets marked for destruction and broken down.
Here, the target is the oestrogen receptor, which drives a common form of breast cancer. The drug treats tumours in which the receptor has mutated to stay permanently switched on and grows despite hormone therapy. In its phase 3 trial, it roughly doubled the time before the cancer progressed compared to the existing drug fulvestrant.
The approach could in principle reach the many disease-causing proteins that have no good pocket to block, which have long been considered ‘undruggable.’ This is just the first approved PROTAC, and many more are likely to come through the pipeline, and change the trajectory of diseases that we consider untreatable today.
What’s behind so much progress?
Part of it is timing: many cancer papers are published to coincide with the American Society for Clinical Oncology’s annual conference. But there’s more beneath that too.
Many new breakthroughs were precision drugs, targeted towards particular genetic mutations and tumours with certain genetic markers. Although the genomic revolution began a few decades ago, some of those advances have taken a long time to make it through the pipeline.
Another part is advances in structural chemistry. Daraxonrasib (the pancreatic cancer breakthrough), enlicitide (the oral PCSK9 cholesterol drug), and retatrutide (the new weight loss drug), for example, came from better understanding of the precise structure of proteins and how they interact with each other.
Both the genomic revolution and structural chemistry have helped researchers understand drug targets better and engineer them, including producing mutant KRAS proteins to find new druggable pockets for the pancreatic cancer drugs, and adapting new platforms, like the mRNA cancer vaccines, for melanoma.
This progress seems like an odd contrast with the cuts we’re seeing to science in the US, including to the National Cancer Institute, which is spending 80 percent less this year than its typical pace. It was responsible for funding fundamental research that eventually led to some of the breakthroughs in this post, including 1980s research into KRAS, which led to new pancreatic cancer drugs, and 2010s research into cancer immunotherapy targeted towards neoantigens, which led to cancer vaccines. The National Heart, Lung, and Blood Institute is also spending around 80 percent less than usual; decades ago, it funded fundamental research on LDL-receptor biology and PCSK9 that brought about cholesterol drugs.
In other words, the breakthroughs we’re seeing today are a lagging indicator of inputs into the pipeline: they reflect choices people made in the past. We’ll only be able to see the gaps created by reduced investment in retrospect, years from now, and see that technology isn’t the only reason for progress; sometimes it’s about the funding, institutions and incentives we create.
Illustration: Bacteriophages at work.
{kind=link}
Hugely interesting. With a recent cancer diagnosis, I really value information like this. E.g. it's probably not new science but in the last few days, NHS England announced it's able to cut radiotherapy sessions for prostate cancer from 20 to 5, saving a lot of appointment time that can be used for other patients. Thanks for this newsletter, it's much appreciated.
You need to slow down or break these up, I can only have my mind blown so many times in one article! But seriously, thanks very much for this amazing update. What a time to be alive!